Первичные, вторичные, третичные алкильные субстраты
Алкильные субстраты способны реагировать, используя разные механизмы:
- Первичные алкильные субстраты взаимодействуют по $SN2$-механизму.
-
Вторичные алкильные субстраты идентифицировать по типу механизма однозначно нельзя. Механизм можно отнести как к классическому $SN2$ или как $SN1$, проходящий через контактную ионную пару.
Например, вторичное алкильное соединение может дать продукт сольволиза с почти полной инверсией, если в качестве растворителя взята муравьиная кислота, проявляющая слабые нуклеофильные свойства.
При вторичных алкильных субстратах возможно частичное образование разделенных ионных пар, так как наблюдается частичная рацемизациия.
-
Третичные алкильные субстраты всегда реагируют по одному из $SN1$-механизмов.
Идентификация классического SN2-механизма и SN1-механизма, проходящего через контактную группу
Измерение относительной стабильности карбокатионов в суперкислых средах и газовой фазе показывает, что ионизация третичных алкильных субстратов должна протекать в $10^9-10^{12}$ раз быстрее, чем ионизация вторичных субстратов.
При сольволизе 2-адамантил- и 2-метил-2-адамантилбромидов в водном растворе этанола или в уксусной кислоте, скорость реакции будет в $10^8$ раз выше скорости сольволиза вторичного субстрата.
Вторичная 2-адамантильная система вступает в реакцию по $SN1$-механизму, проходящему через контактную группу. Стадия определяет скорость ионизации до ионной пары, которая включает 2-адамантильный катион. Этот катион не сольватирован по катионному центру нуклеофильным растворителем, хотя уходящая группа способна сольватироваться через образование водородной связи:
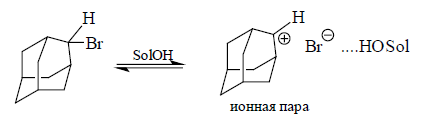
 Найти эксперта
Найти эксперта


В $SN2$-реакцию азид-ион с 2-адамантильными субстратами не вступает, поэтому слабо нуклеофильные молекулы растворителя могут образовывать ионную пару в переходном состоянии.
Влияние нуклеофильности и ионизирующей силы растворителя на сольволиз
В ряду метильные субстраты – этильные субстраты – вторичные и некоторые первичные субстраты – 2-адамантильные системы влияние нуклеофильности растворителя постепенно уменьшается. То есть происходит переход сольволитического механизма от истинного $SN2$-механизма через нуклеофильно сольватированную ионную пару к адамантильной ионной паре.
Нуклеофильное взаимодействие молекул растворителя с электрофильными атомами будет постепенно уменьшаться при переходе от одного субстрата к другому.
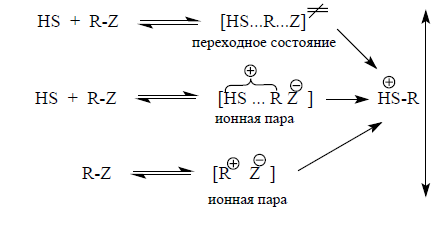
Первая реакция отображает классический $SN2$-механизм. Вторая отображает промежуточный механизм. Третья реакция протекает по $SN1$-механизма, проходящего через контактную группу. $HS$ является молекулой протонного растворителя.
Растворитель выполняет роль нуклеофильного агента. Решающую роль в уменьшении нуклеофильного взаимодействия играют пространственные препятствия, особенно проявляющиеся у 2-адамантильных соединений.
Реакции нуклеофильного замещения алкилгалогенидов
При действии на первичные алкилгалогениды различных полярных или ионных растворителей, содержащих неподеленные электронные пары, происходит замещение галогена. Эти реакции имеют ряд характеристик:
- Скорость реакции будет зависеть как от концентрации субстрата, так и нуклеофила (при соизмеримом соотношении количеств реагентов). В стадии, лимитирующей скорость всей реакции, участвуют и субстрат и нуклеофил.
- Энергия активации является относительно невысокой, поэтому образованию связи $C-Y$ не должен предшествовать разрыв связи $C-Hal$ в субстрате, для которого необходимы большие затраты энергии.
- Если в качестве субстрата брать один из энантиомеров алкилгалогенида, то при замещении в нем атома галогена нуклеофильным реагентом происходит обращение конфигурации. Нуклеофилу более выгодно атаковать молекулу субстрата со стороны, противоположной, замещаемому атому галогена.
Реакции нуклеофильного замещения для третичных алкилгалогенидов несколько отличаются от реакций первичных и вторичных алкилгалогенидов.
При действии на трет-алкилгалогениды солей азотистой кислотыобразуются не нитросоединения, а алкены и эфиры азотистой кислоты; при действии цианидов металлов – не нитрилы, а алкены и изонитрилы и т.д.
Атомы водорода метильных групп, связанные в алкилгалогениде с центральным атомом углерода, способны частично принимать на себя положительный заряд. Чем больше метильных групп связано с центральным атомом углерода, тем больше «распыляется» положительный заряд по периферийным атомам водорода, тем меньше сохраняется его на центральном атоме углерода и тем меньше притягивается к нему атом галогена.
Для третичных алкилгалогенидов гетеролиз молекулы с последующим образованием карбокатиона и галогенид-иона не требует большой затраты энергии, что подтверждается возрастанием дипольных моментов при увеличении числа метильных групп у центрального углеродного атома.
Гетеролиз метилбромида на ионы энергетически невыгоден, так как образуется богатый энергией карбокатион, в котором вероятность рассредоточения положительного заряда мала. Энергетически более выгодно образование трет-бутил-катиона, так как заряд может рассредоточиться на трех метильных группах, в состав которых входят девять атомов водорода.
Устойчивость карбокатионов и выгодность их образования возрастают в ряду:
$C^+ H_3$
Скорость гидролиза третичного алкилгалогенида зависит от его концентрации и не зависит от концентрации гидроксид-ионов в водных растворах. Реакции будут протекать с той же скоростью и при отсутствии щелочи.
В этих реакциях реализуется $SN1$-механизм. Реакция протекает в две стадии:
- гетеролиз алкилгалогенида с образованием карбокатиона; стадия медленная, лимитирует скорость всей реакции;
- карбокатион практически мгновенно взаимодействует с нуклеофильным реагентом или с растворителем.
Скорость реакции имеет нулевой порядок относительно нуклеофильного реагента и первый порядок относительно – алкилгалогенида.
Существование $SN1$-механизма подтверждено фактами:
- сравнительно высокие значения энергий активации, возникающие при гетеролитическом разрыве связей;
- независимость скорости реакции от концентрации нуклеофильного реагента;
- практически полная рацемизация.
Найти эксперта