При переходе от реакций проходящих в газовой фазе к реакциям проходящих в полярных растворителях скорость типичного $SN_2$ процесса уменьшится на 10—20 порядков, например:
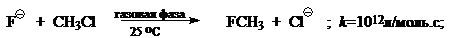
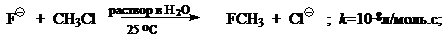
Активность нуклеофила в газовой фазе и в растворителе могжет быть различной, а также могжет изменяться в зависимости от растворителя и их типов. Например, рассмотренный для газовой фазы ряд:
$I^- > Br^- > Cl^- > F^-$
обращается при переходе к протонным растворителям, однако в апротонном растворителе нуклеофильность в этом ряде будет такой же, как и в газовой фазе.
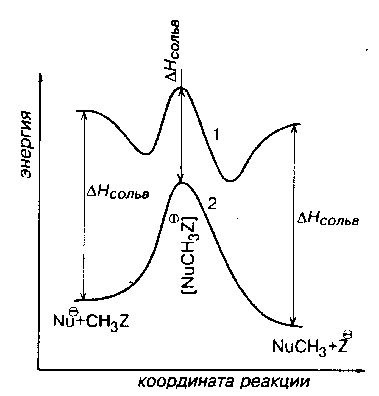
При переходах от реакций в газовой фазе к полярном растворителям происходят очень характерное изменения энергетических профилей $SN_2$-реакций. Потенциальная кривая сглаживается и приобретают привычный профиль, изображенный на рис. 1. (кривая 2).
Рис. 1. Изменения энергетических профилей при переходе от газовой фазы (1) к полярным растворителям (2)
Это означает, что в полярных растворителях образование электростатического комплекса не происходит.
Причины, обусловливающие изменение энергетического профиля
Рассмотрим основные закономерности, которые обусловливают изменения энергетических профилей при переходах от газовой фазы к растворам.
Первая причина «сглаживания ям» состоит в том, что в переходных состояниях реакций электрические заряды делокализованы, а в исходных и конечных точках кривой они локализованы на нуклеофиле Nu или уходящей группе $Z$. Следовательно, энергии сольватации переходных состояний значительно меньше энергий сольватаций анионов с локализованными зарядами, что показано на рис. 1.
Второй причиной изменений форм кривых является то, что реакции нуклеофилов с субстратами в растворе должны предшествовать хотя бы частичные десольватации нуклеофилов и субстратов, приводящие к высвобождениям некоторых количеств молекул растворителя.
Допустим, что на первых стадиях $SN_2$-реакций вместе с частичной десольватацией происходит образование ион-молекулярных комплексов:
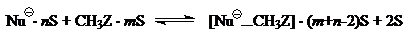
($S$ — молекула растворителя).
Тепловые эффекты этих стадии складываются из эндотермическоих эффектов десольватации ($\triangle H_{сольв}$ на рис. 1) и экзотермических эффектов образований ион-молекулярных комплексов.
Экспериментальные данные и данные расчета показывают, что теплоты десольватаций по абсолютным величинам значительно превышают энергии образований комплексов. Поэтому энергетические «ямы» становятся незаметными, а потенциальная кривая для следующей, собственно химической, стадии сглаживается, приобретая обычный вид.
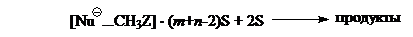
Исходя из этого можно сделать вывод, что энергетический расчет $SN_2$-реакции в газовой фазе в приближении изолированных молекул недостаточен для описания реакции в растворе.
Необходимо учитывать изменение геометрии потенциальной кривой (рис. 1). В областях переходных состояний изменение несущественно. Однако в областях начальных состояний изменения кривых настолько велики, что можно говорить о резком изменении механизма $SN_2$ при переходе из газовой фазы в полярную жидкую среду.
Выбор фазы протекания $SN_2$-реакций
По механизму алифатического нуклеофильного замещения проходят следующие реакции:
- гидролиз алкилгалогенидом;
- гидролиз гемм-дигалогенидов;
- гидролиз 1,1,1-тригалогенидов;
- гидролиз алкильных эфиров неорганических кислот;
- гидролиз диазокетонов;
- гидролиз ацилгалогенидов;
- гидролиз ангидридов;
- гидролиз сложных эфиров;
- гидролиз амидов;
- декарбоксилирование карбоновых кислот и сложных эфиров;
- алкилирование алкилгалогенидами;
- алкилирование ангидридами неорганических кислот;
- дегидратация спиртов;
- переэтерификация простых эфиров;
- алкоголиз ацилгалогенидов;
- алкоголиз ангидридов;
- этерификация кислот;
- переэтерификация;
- образования тиолов, образование сульфидов;
- алкилирование аминов.
И во всех этих реакциях, выбирая в какой фазе их производить, можно регулировать выход того или иного продукта и весь процесс в целом.
Рассмотрим некоторые примеры таких процессов нуклеофильного замещения, где выбор фазы играет решающую роль.
Этерификация кислот щелочами
$RCOOH + R'OH -> RCOOR' +H_2O$
Этерификация кислот щелочами представляет собой реакцию, обратную реакции гидролиза сложных эфиров, и ее удается осуществить только тогда, когда удается сместить равновесие вправо.
Для достижения этой цели используют:
- Использование избытка одного из продуктов (обычно спирта);
- Вывод эфира или воды из сферы реакции;
- Азеотропная отгонки воды.
Если R-метил, тогда равновесие смещают придачу избытка метанола, если $R$-этил, тогда предпочтение отдается азеотропной отгонке воды. В качестве катализаторов используются серная кислота или сульфокислоты бензольного ряда. В случае использования активных кислот катализатор не требуется.
Эту реакцию проводят в жидкой фазе, потому то так легче отгонять мешающую реакции воду.
Обмен галогенов. Реакция Филькенштейна
Обмен галогенов, который иногда называют реакцией Филькенштейна, является равновесным процессом, но достаточно часто равновесие удается сместить в необходимом направлении.
Наиболее широко эта реакция используется для получения фторидов и йодидов. Йодид получают из хлоридов или бромидов. Реакцию проводят в жидкой фазе и как растворитель используют ацетон, в котором йодид натрия растворимы, а хлорид и бромид натрия нерастворимый. Поскольку здесь действует механизм $SN_2$, то для первичных галогенидов реакция идет лучше, чем для вторичных и третичных.
По этой реакции можно не только заменять один галоген на другой, но и осуществлять изотопный обмен с целью получения меченых соединений, которые используют для изучения механизмов реакций.
Алкилирование аминов
$3RX + HN_3 -> R_3N$
$2RX + R'NH_2 -> R_2R'N$
$RX + R'R''NH -> RR'R''N$
$RX + R'R''R'''N -> RR'R''R'''N^+X^-$
Реакция между алкилгалогенидами и аммиаком или первичными аминами конечно непригодна для синтеза первичных и вторичных аминов, так как они являются более сильными основаниями чем исходный аммиак, и сами преимущественно атакуют субстрат. Но эти реакции полезны для получения третичных аминов и четвертичных аммониевых солей. При использовании первичных, вторичных и третичных аминов можно получить соединения, в которых атом азота соединен с различными алкильными заместителями.
 Найти эксперта
Найти эксперта