Полная рацемизация продукта реакции
Понять механизм реакций мономолекулярного замещения позволяет изучение стереохимии данного процесса.
Конечным результатом диссоциативного $SN1$-механизма должна быть полная рацемизация продукта реакции. Это возможно при условии, что и одна, и другая сторона свободного карбокатиона доступны для атаки нуклеофила.
В противном случае, когда карбокатион сольватирован несимметрично, образуется оптически активный продукт, который частично имеет обращенную конфигурацию, а частично является рацемизаванным.
При ацетолизе $n$-бромбензолсульфоната дейтерированного $n$-бутанола происходит 100% инверсия конфигурации и реализуется $SN2$-механизм. Оптически активный субстрат $n-C_3H_7C^*HDOB6$, условием сольволиза является присутствие уксусной кислоты, температура $99 ºС.$
Частичная рацемизация и неполная инверсия продуктов реакции
По одной из версий, частичная рацемизация и неполная инверсия возможны, когда в ходе реакции реализуются два механизма $SN1$ и $SN2$. Однако практически это предположение не подтвердилось.
По другой версии считается, что частичная рацемизация возможна при различии в стабильности карбокатионов. При этом менее стабильный карбокатион реагирует очень быстро, даже если атака нуклеофила происходит в основном с тыльной стороны и одна из его сторон экранирована уходящей группой.
Например: У соединения $C_6H_5C*HDO-O_2SC_6H_4CH_3-n$ в процессе сольволиза наблюдается неполная инверсия (90%) и частичная рацемизация (20%) (в присутствии уксусной кислоты при температуре 25ºС).
При полной диссоциации оптически активного субстрата образуется симметричный карбокатион. Для него два альтернативных переходных состояния – атака с тыла и фронтальная атака – являются энантиомерными, то есть имеют одинаковую энергию.
В случае частичной рацемизации два переходных состояния обладают разной энергией. Уходящая группа остается связанной с атомом углерода, при этом ахиральный ион не образуется.
На первой медленной стадии ионизации $RZ$ уходящая группа входит в состав переходного состояния и интермедиат нельзя описывать как карбокатион.
Исследования ионных пар С. Уйнстейном
С. Уйнстейн обнаружил, что ионизация органической ковалентной молекулы происходит в ходе нескольких последовательных стадий:
- Вначале образуется «интимная» (контактная) ионная пара $R+Z^-$, в ней катион и анион связаны между собой, разделяющих молекул растворителя нет.
- При разделении компонентов ионной пары «интимная» ионная пара «рыхлую» (сольватно - разделенную) ионную пару $R+ǁZ^-$, в этом случае ионы разделены молекулами растворителя.
- Дальнейшая диссоциация сольватно – разделенной пары на «свободные» сольватированные ионы.
Образуется два вида ионных пар: сольватно – разделенные и контактные, при этом электростатическое взаимодействие в контактной паре будет значительно выше, что определяет высокую реакционную способность «рыхлых» ионных пар.
Электропроводность раствора определяется свободными диссоциированными ионами. Сольватно - разделенные и контактные ионы в этом процессе не участвуют. Существование ионных пар в растворе возможно, если енергия электростатического притяжения частиц с разными зарядами выше энергии теплового движения ионов.
Способность растворителя к ионизации ковалентной молекулы $R-Z$ может быть:
- ионизирующей;
- диссоциирующей.
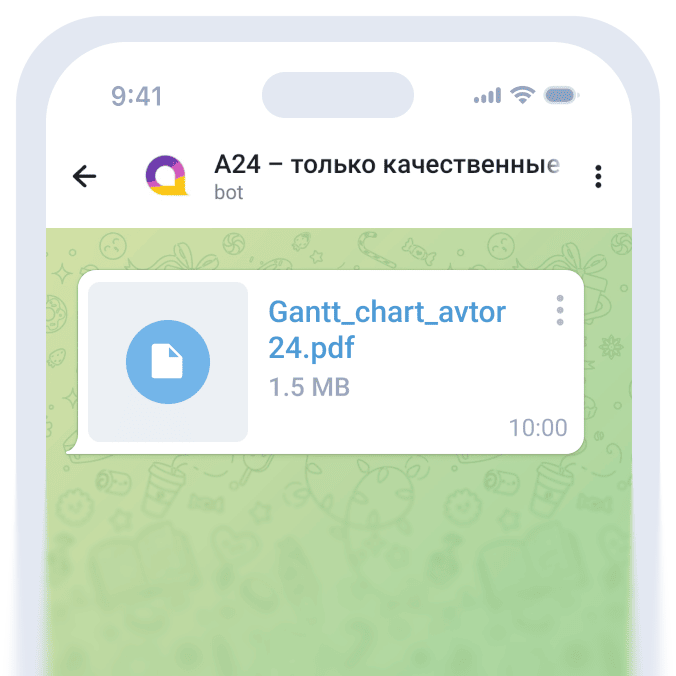
Ионизация и диссоциация трифенилхлорметана (тритилхлорид). В малополярных растворителях (уксусная кислота, пара-крезол) трифенилхлормет ионизирован, но не диссоциирован. Ионизация вызвана образованием прочной водородной связи с уходящей группой:
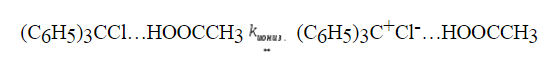
Ацетонитрил и нитробензол обладают высокой диэлектрической проницаемостью. В них трифенилхлорметан не ионизирован. При наличии добавок кислот Льюиса $SbCl_5$, $SnCl_4$, $AlCl_3$ в тринитрохлорметан в ацетонитриле или нитробензоле происходит ионизация и последующая диссоциация субстрата:
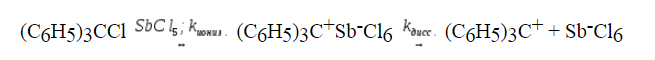
В жидком $SO_2$ трифенилхлорметан частично ионизирован и частично диссоциирован (при 10ºС):
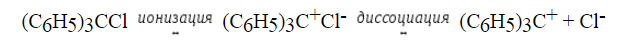
Сольватно – разделенные и контактные ионные пары играют важное значение в процессах $SN1$-замещения у насыщенного атома углерода.
При нагревании (+)-трео-3-анизилбутил-2-брозилата в уксуснокислом растворе происходит сольволиз (замена брозилатной группы на ацетатную) и рацемизация непрореагировавшего субстрата:
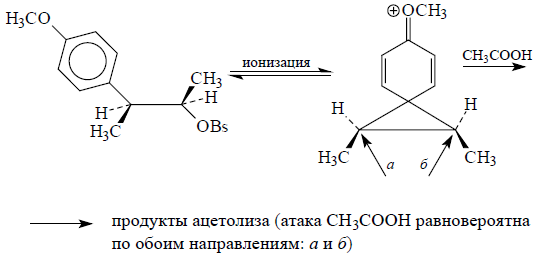
При отщеплении аниона $OBs-$ образуется ахиральный анизилбутильный карбокатион, который имеет мостиковую структуру.
Сольволиз исходного брозилата в уксусной кислоте происходит медленнее, чем рацемизация. За обратимой и быстрой ионизацией исходного брозилата происходит более медленная атака мостикового анизилбутильного карбокатиона молекулой растворителя. Однако, в присутствии солей с общим ионом $OBs-$ сольволиз замедляться не будет.
За основу принят ион – парный механизм, согласно которому образование ионной пары из исходного брозилата является медленной стадией:
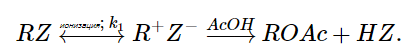
Рацемизация исходного брозилата вызвана быстрым равновесием с участием ионной пары. На обратной стадии «внутреннего возврата» брозилат – анион может с равной долей вероятности атаковать внутри пары оба соседних атома углерода (а и б), что ведет к рацемизации. В этом внутримолекулярном процессе брозилат – ион не становится свободным, следовательно добавки общего иона на скорость сольволиза не влияют. Если в раствор ввести перхлорат лития $LiClO_4$, то наблюдается увеличение как скорости рацемизации, так и скорости сольволиза.
- При большой концентрации перхлората лития кривые нормального солевого эффекта для рацемизации и сольволиза почти параллельны.
- При низких концентрациях перхлората лития наблюдается специальный солевой эффект: крутой подъем кривой зависимости скорости сольволиза от концентраций.
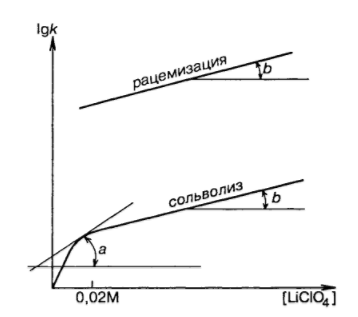
Тангенс угла $b$ показывает нормальный солевой эффект, тангенс угла $a$ – специальный солевой эффект.
С. Уйнстейн предположил, что в процессе ионизации происходит последовательное образование ионных пар: контактной и сольватно – разделенной. При этом контактная ионная пара подвергается рацемизации, а в сольволизе задействованна сольватно – разделенная ионная пара.
Схема, показывающая реакции мономолекулярного нуклеофильного замещения:
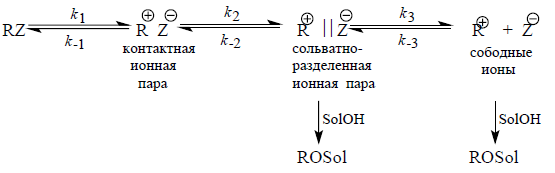
Продукты сольволиза образуются из сольватно – разделенной ионной пары и частично из свободного карбокатиона. Возврат к субстрату $RZ$ может протекать на всех стадиях.
 Найти эксперта
Найти эксперта