




 Найти эксперта
Найти эксперта


Присоединение спиртов и фенолов
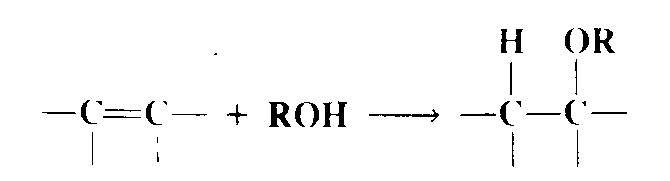
Присоединение спиртов и фенолов к двойным связям катализируется кислотами и основаниями. При кислотном катализе реакция идет по электрофильному механизму, а атакующей частицей является $H^+$. Получающийся карбокатион взаимодействует с молекулой спирта:
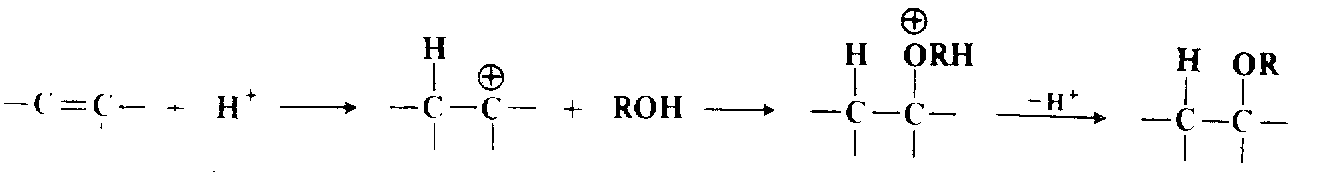
Следовательно, такое присоединение подчиняется правилу Марковникова. Первичные спирты дают результаты лучшие, чем вторичные, а третичные спирты весьма инертны в этой реакции. Реакция представляет собой удобный метод синтеза третичных простых эфиров при использовании подходящего олефина, такого, как $Мe_2C = CH_2$.
С субстратами, более подверженными атаке нуклеофилов, например полигалогеноолефинами и олефинами типа $C=C—Z$, лучше проводить реакцию в щелочном растворе, где атакующей частицей является RO. Реакции с субстратами типа $C=C—Z$ относятся к реакциям присоединения по Михаэлю, и группа $OR$ всегда присоединяется к атому углерода, более удаленному от группы $Z$.
Присоединение карбоновых кислот
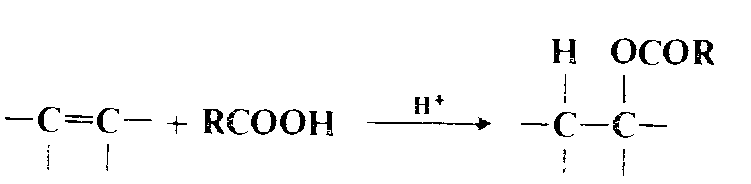
Сложные эфиры карбоновых кислот получаются при присоединении карбоновых кислот к олефинам. Эта реакция обычно катализируется кислотами (протонными или кислотами Льюиса и по механизму аналогична предидущей реакции. Поскольку здесь соблюдается правило Марковникова, то из олефинов типа $R_2C = CHR$ можно синтезировать труднодоступные сложные эфиры третичных спиртов. Наиболее подходящим растворителем для этой реакции является третбутиловый спирт. При обработке сильной кислотой карбоновой кислоты, содержащей углеродуглеродную двойную связь, присоединение происходит внутримолекулярно. А продуктом является лактон независимо от исходного положения двойной связи в цепи, поскольку сильные кислоты катализируют и перемещение двойных связей. Двойная связь всегда мигрирует в положение, удобное для реакции, независимо от того, приближение это или удаление от карбоксильной группы. Но с обсуждаемым процессом конкурирует еще одна реакция, в которой образуется производное циклопентенона или циклогексенона.
Двойная связь сначала мигрирует в удобное для реакции положение, а затем преимущественное протекание реакции по пути а или б зависит главным образом от типа кислоты, используемой в качестве катализатора. Протонные кислоты, такие, как серная, муравьиная и $HF$, дают в основном лактоны, тогда как кислоты Льюиса, например уксусный ангидрид, хлорид цинка в уксусной кислоте, $P_2OS_5$ и т. д., преимущественно дают кетоны. Поскольку интермедиатами являются карбокатионы. то в обеих реакциях наблюдаются перегруппировки.
Присоединение сероводорода и тиолов
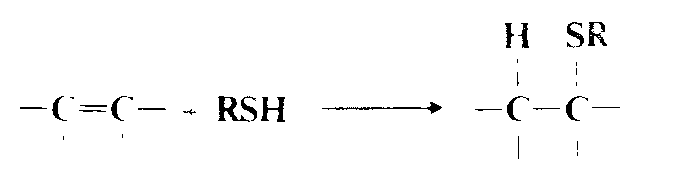
Присоединение сероводорода и тиолов к олефинам может идти по электрофильному, нуклеофильному или свободнорадикальному механизму . В отсутствие инициаторов присоединение к простым олефинам осуществляется по электрофильному механизму, аналогичному механизму предидущей реакции, и правило Марковникова соблюдается. Однако это присоединение обычно происходит очень медленно, и часто в отутствие кислотного катализатора не может быть осуществлено вообще или требует очень жестких условий. Реакцию, например, можно провести в концентрированной серной кислоте . В присутствии свободнорадикальных инициаторов сероводород и тиолы присоединяются к двойным и тройным связям по свободнорадикальному механизму с ориентацией против правила Марковникова . По существу, направление ориентации можно использовать для установления типа реализующегося механизма. В реакции свободнорадикального присоединения можно ввести сероводород, $RSH$ (группа $R$ может быть первичной или третичной), $ArSH$ и $RCOSH$. Фрагмент $R$ может содержать различные функциональные группы. Олефины, к которым идет присоединение, могут быть терминальными и внутренними, иметь разветвления или циклическую структуру, содержать различные функциональные группы, включая $OH$, $COOH$, $COOR$, $NO_2$, $RSO_2$ и т. д. К ацетиленам можно присоединить один или два моля $RSH$.
Первоначальным продуктом присоединения сероводорода к двойной связи является тиол, который способен присоединиться к другой молекуле олефина, поэтому часто получаются сульфиды:
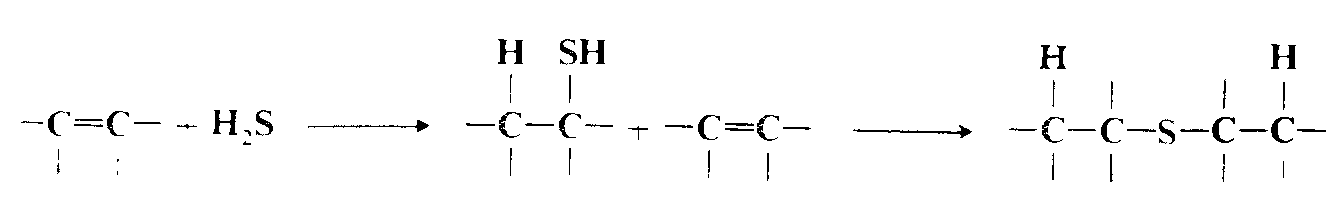
Присоединение бисульфита натрия
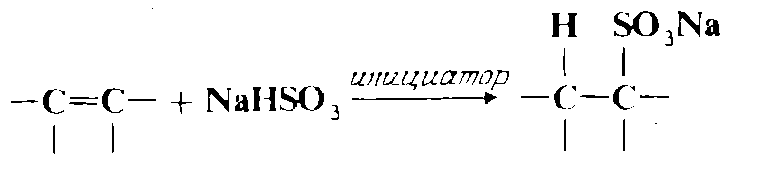
В результате присоединения бисульфитов к олефинам в присутствии свободнорадикальных инициаторов можно получить Фоли алифатических сульфоновых кислот. Присоединение происходит против правила Марковникова.
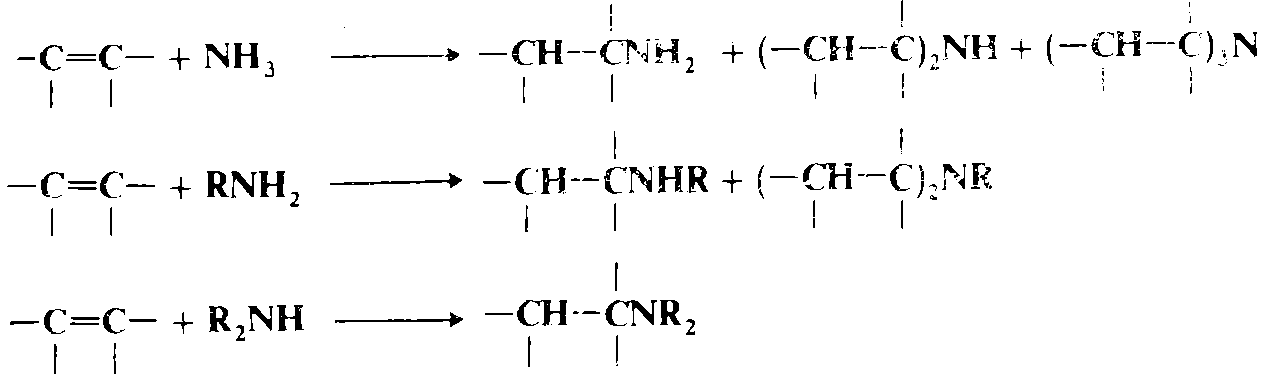
Гидроаминоприсоединение
Аммиак, а также первичные и вторичные амины присоединяются к олефинам, чувствительным к нуклеофильной атаке. В реакции присоединения аммиака возможно образование трех продуктов, поскольку продукт первоначального присоединения представляет собой первичный амин, который может присоединяться ко второй молекуле олефина и т. д. Аналогично из первичных аминов получаются вторичные и третичные продукты. На практике обычно удается добиться преимущественного образования одного продукта. Поскольку аммиак и амины представляют собой значительно более слабые кислоты, чем вода, спирты и тиолы, и поскольку кислоты практически не катализируют эти реакции (так как под их действием $NH_3$ превратится в $ΝH_{4^+}$), то это взаимодействие не может идти по электрофильному механизму, и поэтому с обычными олефинами реакция если и идет, то выходы очень низкие, кроме случаев проведения процессов в очень жестких условиях (например, при 178—200 °С, 800—1000 атм и в присутствии металлического натрия для реакции аммиака с этиленом). Механизм таких превращений почти всегда нуклеофильный, и реакции чаще всего проводят с полигалоге поолефинами , субстратами Михаэля и алкинами. Как и следует ожидать, в случае субстратов Михаэля азот присоединяется к атому углерода, не соединенному с группой $Ζ$. К олефинам присоединяются и другие азотсодержащие соединения, например гндроксиламин, гидразины, амиды ($RCONH_2$ и $RCONHR'$, включая имиды и лактамы), а также сульфамиды. В случае амидов требуется катализ основаниями, поскольку сами амиды — недостаточно сильные нуклеофилы для проведения реакции и их необходимо превратить в $RCONH"$. Основные катализаторы часто используются даже для присоединения аминов так, чтобы действующим нуклеофилом были ионы $RNH$ или $R_2N$. Третичные амины (кроме тех, стерический объем которых слишком велик) присоединяются к субстратам Михаэля по реакции, которая катализируется такими кислотами, как $HCl$ или $HΝO_3$, давая соответствующие четвертичные аммониевые соли.
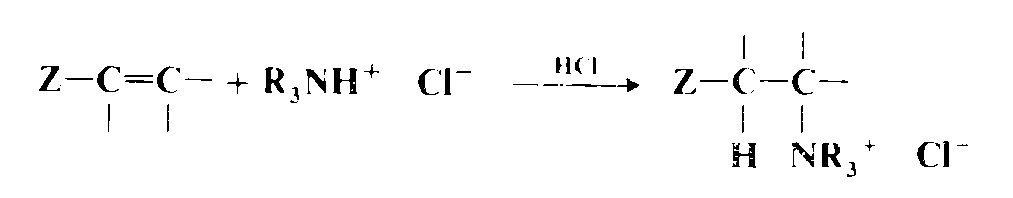
Присоединение аммиака к двойным связям (даже обычным) можно осуществить косвенным путем через гидроборированне с последующей обработкой $NH_2Cl$ или $NH_2OSO_2OH$. Эта методика приводит к первичному амину с ориентацией против правила Марковни кова. Косвенный метод присоединения первичного или вторичного амина к двойным связям заключается в аминомеркуриро вании с последующим восстановлением (аналогичная методика оксимеркурирования — демеркурирования).
Присоединение азотистоводородной кислоты.
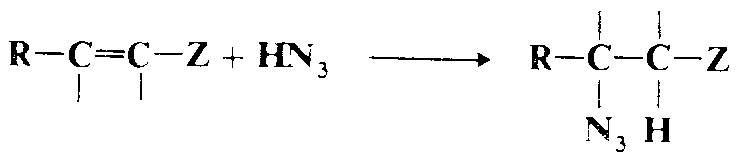
Азотистоводородная кислота присоединяется к определенным субстратам типа $C=C—Z$, давая βазидосоединения . Реакцию не удается провести, если $R$ — фенильная группа. Косвенным путем $HN_3$ присоединяется к обычным олефинам через азидомеркурирование с последующим демеркурированием.
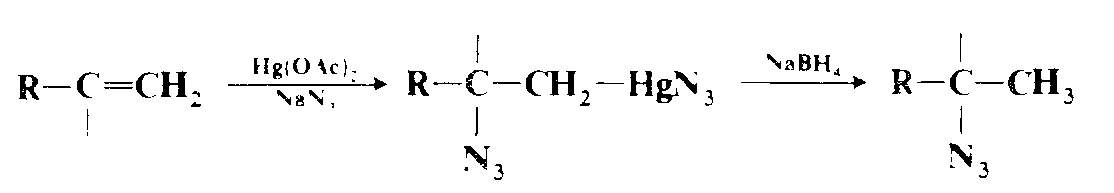
Этот метод применим к терминальны м алкенам и напряженным циклоалкенам, непригоден в случае ненапряженных внутренних алкенов.
Присоединение других электрофильных реагентов
Многие другие электрофильные агенты также присоединяются по двойной связи алкенов. Среди них отметим бромазид $Br+δN_3δ$ и йодазид $I+δN_3δ$, йодизоцианат $I+δ$ $N_3δ = C = O$, диродан, хлористый нитрозил $Clδ – N+δ = O$, $C_6H_5I(OAc)^2$ и $RSeCl$. Присоединение $IN_3$, $BrN_3$, $INCO$ и других псевдогалогенидов, где азид, изоцианат и тиоцианат выполняют функцию галогенидиона, по своему механизму, стерео и региоселективности принципиально не отличается от присоединения несимметричных галогенов. Присоединение $IN_3$, $BrN_3$ и $INCO$ имеет синтетическое значение, поскольку азидогруппа может быть восстановлена до аминогруппы с помощью диборана $B_2H_6$ или каталитически водородом в присутствии палладия на карбонате бария. Сам йодазид получают в растворе в эфире при обработке азида серебра или натрия эфирным раствором йода или хлористого йода при 0 – 10ºС:
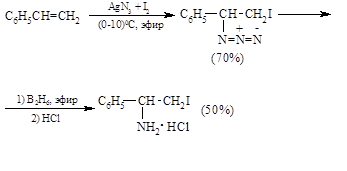 Найти эксперта
Найти эксперта



