




 Найти эксперта
Найти эксперта


В предыдущем разделе интерпретация правила Марковникова строилась на основе сравнения относительной стабильности карбокатионных интермедиатов, образующихся при реакции алкенов с протоном. Другой путь к пониманию правил ориентации присоединения по двойной связи состоит в использовании качественного метода молекулярных орбиталей. Ориентация по правилу Марковникова и против этого правила во многих случаях можно легко предсказать при рассмотрении молекулярных орбиталей исходных алкенов.
Классический механизм электрофильного присоединения, предложенный пятьдесят лет назад, как правило, предполагает присоединение электрофила $E^{(+)}$ по двойной связи с образованием в качестве интермедиата «ониевого иоиа» (в том случае, когда речь идет о присоединении положительно заряженного иона галогена, происходит образование галогенониевого иона). Этот интермедиат представляет собой трехчленный цикл, в котором положительный заряд распределен по трем центрам: атому $E$ и двум атомам углерода. На второй стадии реакции электрофильного присоединения ($Ade$) нуклеофил $Nu^{(-)}$ атакует ониевый ион, причем эта атака осуществляется строго стереоселективно в той полуплоскости, где нет электрофильного заместителя Е, в результате чего образуются продукты анти-перипланарного присоединения, а двугранный угол ($ECC$, $CCNu$) равен 180°.
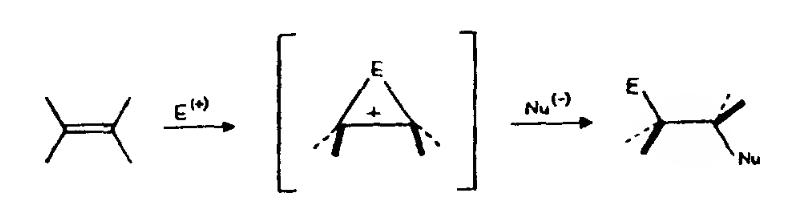
Принято считать, что реакции электрофильного присоединения ($Ade$) С классическим механизмом протекают под орбитальным контролем. В приближении метода граничных орбиталей (метод Фукуи) связывающее электрон-электронное взаимодействие представляет собой перекрывание $HBMO$ электрофила $E^{(+)}$ и ВЗМО олефина, причем последняя представляет собой дважды вырожденный π-уровень. Для того чтобы перекрывание между НВМО электрофила [для простоты рассматривают $ls(H^+)$ или $3p(C_{1^+})]$ и ВЗМО олефина было максимальным, треугольник $ECC$ в переходном состоянии должен быть равносторонним. Это полностью соответствует классическому описанию, где интермедиат является «ониевым ионом». в переходном состоянии должен быть равносторонним. Это полностью соответствует классическому описанию, где интермедиат является «ониевым ионом».
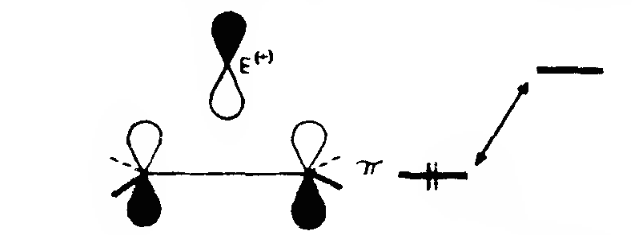
Обычно описываемые реакции проводят в растворе с такой диэлектрической проницаемостью, при которой процесс может протекать как под орбитальным контролем, так и в условиях зарядового контроля. В чистом виде переходное состояние в реакциях электрофильного присоединения представляет собой гибрид, граничными формами которого являются карбокатион и онневый ион. Таким образом, перед нами стандартный механизм Ade-реакции электрофильного присоединения.
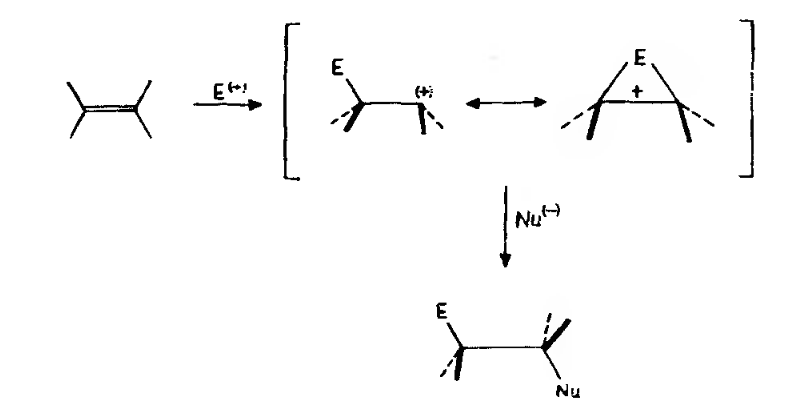
Современная формулировка правила Марковникова позволяет объяснить региоселективность, наблюдаемую в реакциях электрофильного присоединения: преимущественно образуется наиболее замещенный (наиболее стабильный) карбокатион. Правилу Марковникова подчиняется и микроскопически обратимая реакция - синтез трет-бутилхлорида. При присоединении молекулы $HCl$ к изобутилену происходит через образование относительно стабильного трет-бутильного катиона. Среди продуктов реакции не удалось обнаружить даже следов 1-хлор-2-метилпропана, образование которого должно происходить через изо-бутильный катион.
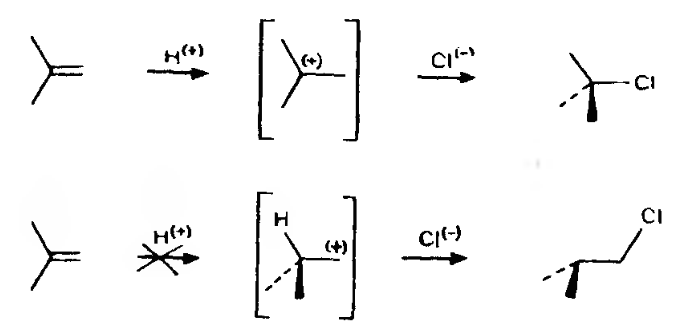
Объяснение механизма реакции гидрогалогенирования представлением об орбиталях
Механизм реакции гидрогалогенирования объясняется представлениями об орбиталях: заместитель при двойной связи изменяет соотношение атомных орбиталей, участвующих в образовании π-связывающей молекулярной орбитали (МОСВяз) и разрыхляющей молекулярной орбитали (МОразр). Так. в пропене метальная группа, обладающая положительным индуктивным эффектом ( + $I$-эффект), увеличивает долю участия $AOC_1$ по сравнению с $AOC_2$ В МОсвяз И одновременно увеличивает участие $AOC_2$ и уменьшает участие $AOC_1$ в МОразр (размеры объемных восьмерок означают долю участия атомных орбиталей образовании молекулярных орбиталей):
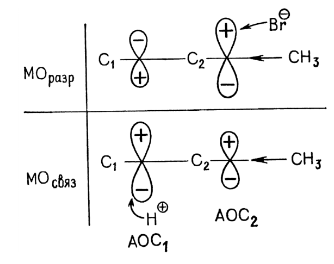
Поэтому электрофильный реагент (протон) будет стремиться занять положение, наиболее близкое к месту наибольшей концентрации заряда на МОсвяз, и, следовательно, атаковать атом $C_1$. Нуклеофильный реагент будет взаимодействовать с $AOC_2$ разрыхляющей орбитали и атаковать атом $C_2$.
Отклонение от правила Марковникова
Американский ученый М. Хараш в 1938 г. обнаружил, что в присутствии перекисей — источников свободных радикалов , например метальных $CH_3$, галогеноводородные кислоты присоединяются не по правилу Марковникова (перекисный эффект Хараша). Причина заключается в том, что изменение условий реакции изменяет ее механизм, который становится радикальным, и образуются иные конечные продукты.
Инициирование реакции:
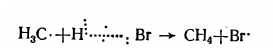
Цепная реакция:
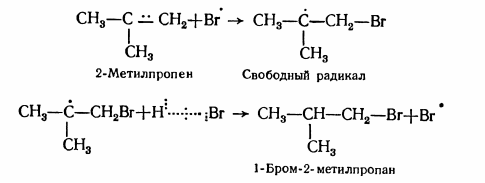
Под влиянием атомного брома происходит разобщение $\pi$-электронов 2-метилпропена, который образует ковалентную связь с бромом и превращается в свободный радикал. Последний, реагируя с бромоводородной кислотой, стабилизируется затем в 1-бром-2- метилпропан. Обратный порядок присоединения галогеноводородной кислоты к 2-метилпропену приведет к возникновению свободного радикала с одиночным электроном у крайнего атома углерода:
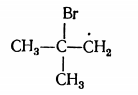
Порядок присоединения галогеноводородных кислот к двойной связи зависит от различных факторов:
-
Во-первых, как и в случае ионного гидрогалогенирования, это объясняется устойчивостью свободного радикала. Атомный галоген атакует тот атом углерода при двойной связи, в результате присоединения к которому образуется наиболее устойчивый промежуточный радикал. Устойчивость радикала растет с увеличением числа групп, связанных с радикальным атомом углерода. Причина этого явления, по-видимому, заключается в увеличении возможности сопряжения свободного электрона с электронными облаками соседних $C—H$-связей и, следовательно, в возрастании степени его делокализации:
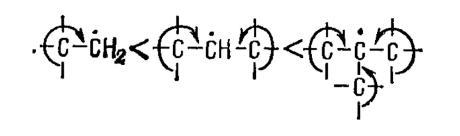
Естественно, что активность свободных радикалов изменяется в обратном порядке.
-
Во-вторых, имеют значение стерические факторы: радикал атакует пространственно наиболее доступный атом углерода. Поэтому в рассматриваемой реакции образуется третичный свободный радикал. Одиночный $p$-электрон занимает атомную орбиталь ($p-AO$), а три σ-связи трехвалентного атома углерода образованы $sp2$-гибридными орбиталями:
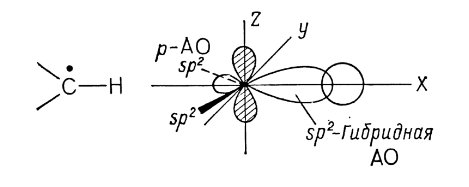
Найти эксперта



