




 Найти эксперта
Найти эксперта



Электрофильные реакции алканов хоть и мало используются в лабораторных синтезах, но они, как уже сказано выше, широко используются в промышленности. Это - каталитические процессы крекинга нефтяных фракций, алкилирования изопарафиновых и ароматических углеводородов олефинами, изомеризации нормальных парафинов, ароматизации парафиновых углеводородов. Все эти процессы реализованы на кислотных катализаторах: крекинг, изомеризация и ароматизация - на твердофазных, алкилирования - на жидкофазных (смесях высококонцентрированных серной и фтористоводородной кислотах).
Закономерности каталитических процессов крекинга нефтяных фракций
Основой механизмов протекания реакций катализируемых кислотными цеолитами является классическая карбонионная теория, в рамках которой постулирована перемещение водорода и алкильных групп вдоль парафиновых цепочек. Хотя доминирующим остается представление о высочайшей стабильности третичных карбокатионов, однако начинают появляться работы, которые допускают преобразования третичного карбокатиона во вторичный, а также исследования, согласно которым в ряду "третичные – вторичные - первичные" карбокатионы наиболее реакционные признаны первичные. Несомненным является возникновение метильного разветвления преимущественно при втором атоме углерода в случае изомеризации легких н-парафинов.
Все сильнее утверждается суперкислотная теория Ола, в соответствии с которой реально протонирование молекул метана и этановых метильных групп на суперкислотах и цеолитах. В стабилизации образованных неклассических первичных карбокатионов на цеолитах решающую роль играют отрицательно заряженные позиции цеолитных структур. При распаде неклассических карбокатионов, по теории Ола, заряд может оставаться как в своей начальной позиции, так и сдвигаться к соседнему атому углерода. Это дает две разные пары "парафин-олефин". Одним из самых ярких новых экспериментальных фактов является способность к дейтерированию первичных атомов углерода на кислотных формах цеолитов и полной неспособности к этому третичных углеродных атомов.
Ключевыми позициями в этих процессах являются:
- Протонирование насыщенных структур на цеолитных катализаторах по третичным атомам углерода, как и дейтероообмен водорода при этих атомах на цеолитах, следует считать маловероятным из-за стерической недоступности данных атомов как для дейтерия, так и для протонов;
- Вероятным механизмом активации парафиновых углеводородов на кислотных цеолитах, как и дейтероообмен, является их протонированин по метильным группам как стерически доступных для непосредственного воздействия протонов $\beta$-центров;
- Протон неклассического карбокатиона может перемещаться вдоль парафиновых цепочек в других позиций, а именно - второго и третьего атомов углерода. При этом, чем сильнее $\beta$ центр, тем более отдаленным может быть перемещение;
- Неклассические карбокатионы могут распадаться или переходить в классические частицы предварительно потеряв два атома водорода;
- Акцептором водорода, является $L$-центры катализатора;
- Как неклассические, так и классические первичные карбокатионы стабилизируются отрицательно заряженными позициями кристаллической решетки цеолита;
- Классические карбокатионы распадаются по правилу разрыва С-С-связей
- На завершающих стадиях реакции $L$-центры десорбируют диссоциативный (в виде протона и гидрид иона) хемосорбований водород, чем обеспечивают последнюю стадию механизма реакции с образованием нейтральных продуктов превращения и восстановлением β-центров катализатора;
- -торичные и третичные карбокатионыиз стерических причин не могут получить гидрид-ион - их могут получить только первичные карбокатионы.
Примеры электрофильных реакций не имеющих промышленного значения
-
Водородный обмен
Этот тип реакций частично уже рассматривалась выше. Легкий обмен водорода в положении 1 адамантана в суперкислой среде является доказательством треугольного переходного состояния:
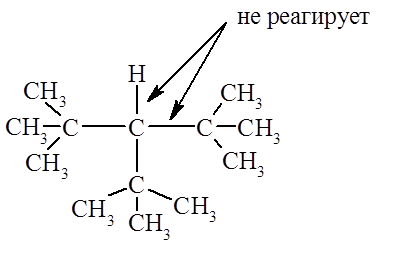
В парафинах в растворах $\frac{FSO_3H}{SbF_5}$ при низких температурах (-80ºС) преобладает расщепление связей $C – H$, а при высоких температурах – связей $C–C$. Пространственные препятствия могут влиять на протекание процесса очень сильно. Так, в 3-трет-бутилметане третичные связи очень сильно экранированы, и поэтому реагируют лишь $C–CH_3$ и первичные $C – H$-связи:
-
Алкилирование
Электрофильное алкилирование осуществляется или при взаимодействии алканов с заранее полученным карбокатионом структуры $R_3C^+$ (например, $t^-Bu+SbF_{6^-}$), или при реакции алкана с карбокатионом $R3C^+$, который возникает в результате переноса протона (например, $R_3CH + H^+ = H_2 + R_3C^+$).
Реакции алканов со стабильными карбокатионами обычно изучают в растворае сульфурилфторидхлорида при -78ºС например:
Уменьшение пространственных препятствий повышает выход:
-
Нитрование
Чтобы избежать свободнорадикального механизма при нитровании и кислотного расщепления продуктов реакции, для нитрования используют не саму азотную кислоту, а стабильные соли нитрония (например, $NO_2+PF_{6^-}$) в апротонном растворителе (например, смеси хлористого метилена с тетраметиленсульфоном). В случае метана и этана продукты нитрования - нитрометан и нитроэтан –не чувствиетельны к кислотам и в качестве растворителей можно применять безводную $HF$ и $FSO_3H$, в которых выходы выше
-
Хлорирование
При электрофильном хлорировании алканов выход продуктов изменяется от 2 – 5% (для метана) до 55 – 60 (для высших алканов). Особенность реакции электрофильного галогенирования состоит в ее высокой селективности: из метана образуется только монопродукт $CH_3Cl$ и не образуются дизамещенный $CH_2Cl_2$, и другие хлорметаны.
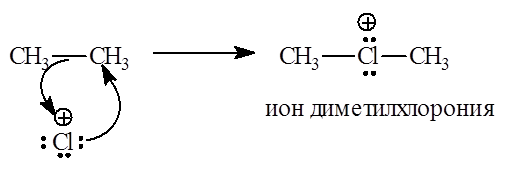
Промежуточными частицами в реакции «$Cl^+$» со связью $C–C$ алканов
являются диалкилхлорониевый ион с открытой цепью:
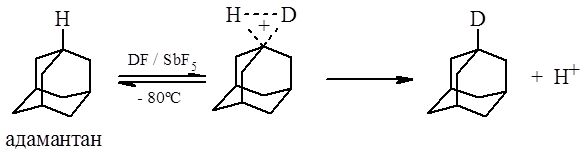
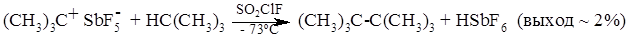
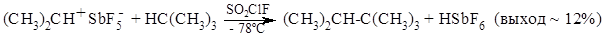
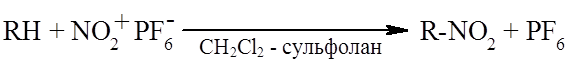
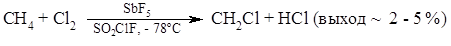
Перегруппировки
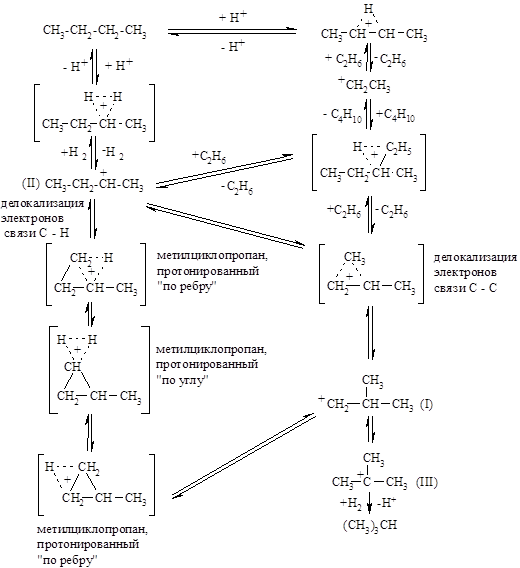
В процессе изомеризации алканов, катализируемом кислотами, первичным актом является присоединение протонов с образованием пятикоординационных катионов. В последующей стадии при отщеплении молекул водорода или низкомолекулярных алканов образуются трехкоординационные катионы, которые можгут перегруппировываться в более стабильные катионы путем разного рода внутри- и межмолекулярных процессов (алкилирование, отщепление алканов или молекул водорода, перемещение алкилов или атомов водорода). На приведенной ниже схеме показаны процессы изомеризации н-бутана в изобутан в кислой среде. В этой сложной реакции участвуют первичный (I), вторичный (II) и третичный (III) бутильные катионы, а также метилциклопропаны, протонированные «по ребру» или «по углу». Все приведенные реакции обратимы, но равновесие очень сильно смещено в сторону трет-бутильного катиона (III), так что в спектрах ЯМР обнаруживаются лишь сигналы от этого катиона, но нет сигналов от катионов (I) и (II), которые, вероятно, образуются лишь в очень малой концентрации:
 Найти эксперта
Найти эксперта


