




 Найти эксперта
Найти эксперта



Сегодня общепринято, что простейшую газофазную реакцию описывают двухъямной потенциальной кривой. Такая кривая для реакции
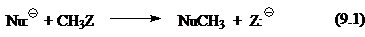
изображена на рис. 1. Левому минимуму соответствует предреакционный комплекс $Nu^-...CH_3Z$, а правому — послереакционный комплекс $NuCH_3...Z^-$. Энергии предреакционных комплексов ниже, чем энергии реагентов $(Nu^- + CH_3Z)$, а энергии послереакционных комплексов ниже энергий продуктов реакции $(CH_3Nu + Z^-)$. Это связано с тем, что свободный ион в газовой фазе исключительно нестабилен. Связ, обозначенная в комплексе пунктиром, имеет в основном элек-тростатическую природу, при этом во фрагменте $CH_3Z$ или $NuCH_3$ связь почти не искажена и имеет в основном ковалентный характер. Превращения предреакционных комплексов в послереакционные происходит через седловую точку на кривой. Этой точке отвечают переходные состояния $[Nu ... CH_з... Z]^-$ с более или менее симметричными структурами (в зависимости от $Nu:^-$ и $Z$) и делокализованными зарядами.
Образование ион-молекулярного комплекса было установлено экспериментально. В газовой фазе получен комплекс типа $Hal^-...CH_зHal$, энергия электростатической связи в котором составляет от 8,6 до 14,4 ккал/моль. Этот комплекс в принципе аналогичен комплексу с водородной связью $B^-....HA$, который образуется при переносе протона между кислотой ($HA$) и основанием ($B^-$) Бренстеда.
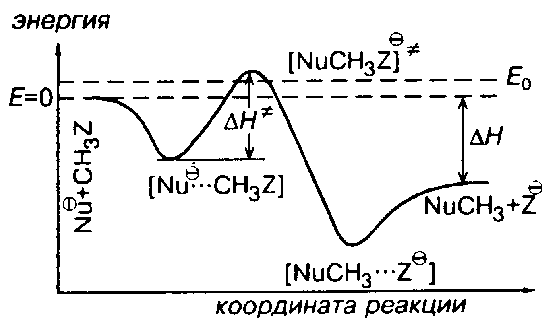
Рис. 1. Энергетический профиль газофазной реакции, протекающей по $SN2$ механизму. $Е0$ - кинетическая энергия реагентов; $ΔH$ - тепловой эффект реакции; $ΔH$ - энтальпия активации
Кинетика экзотермического процесса
Кинетика экзотермических процессов с двухъямными профилями может быть описана следующей схемой:
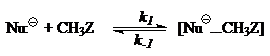
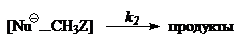
Применяя принципы стационарности, для констант скорости получим соотношение:
$k_{набл}$=
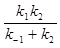
Исходя из чего, значения kнабл в общем случае зависят как от скоростей образования, так и от скоростей распада предреакционных комплексов. Если $k_{-1}$ >> $k_2$ , т.е. скорости определяются стадиями образования комплексов $[Nu^-....CH_3Z]$. Обычно для реакций протекающих по $SN2$ механизму в газовой фазе наблюдается первый случай, когда $k_{-1} >> $k_2$, и скорость замещения определяется тогда как стадиями образования, так и стадиями распада предреакционных комплексов., и скорость замещения определяется тогда как стадиями образования, так и стадиями распада предреакционных комплексов.
Факторы, от которых зависит реакционная способность
В таком случае реакционные способности зависят одновременно и от природы нуклеофила $Nu^-$, и от природы уходящей группы $Z$. Из-за этого единые «шкалы нуклеофильности» анионов $Nu^-$ или «шкалы нуклеофугности» анионов $Z$ трудно создать для газофазных процессов. Тем не менее в ряде газофазных реакций, таких как:
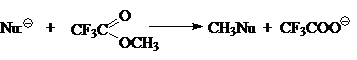
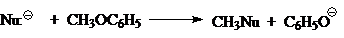
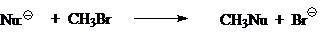
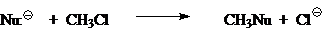
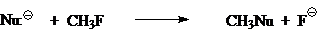
за некоторым исключением, качественный ряд изменения силы нуклеофилов $Nu:^-$ выглядят одинаково. Для таких реакций ряд изменения силы нуклеофилов будет выглядеть так:
$НО^- > F- > СН_3О^- > СН_3S- > CN^- ~ Сl^- > Вr^-$.
Этот ряд нуклеофильности примерно соответствует изменению кислотности $Nu^-H$ в газовой фазе (за исключением того, что $F^-$ и $СН_3О^-$ поменялись местами).
Несмотря на существование отдельных корреляций между кислотностью и нуклеофильностью, в целом скорость $SN_2$-реакции не коррелирует с кислотностью нуклеофила, т.е. более соединение с более сильной кислотностью не обязательно должно быть более слабым нуклеофилом.
α-Эффект в нуклеофильном реагенте
Анионы или нейтральные молекулы, имеющие атом с неподеленной парой электронов, который непосредственно связан с нуклеофильным центром, проявляют свойства гораздо более сильных нуклеофилов, чем их аналоги, не содержащие такого атома по соседству с реакционным центром. Так например, гидразин $NH_{2^-} NH_2$ или гидроксиламин $NH_2OH$ значительно более сильные нуклеофилы, чем аммиак или первичные амины, так же как пероксид-ион и гипохлорид значительно более сильные нуклеофилы по сравнению с $НО^-$. Такое особое поведение этих нуклеофилов получило название $α$-эффекта.
Нуклеофильная реакционная способность и основность реагентов изменяются параллельно, если они имеют один и тот же атом с неподеленной парой электронов. Например:
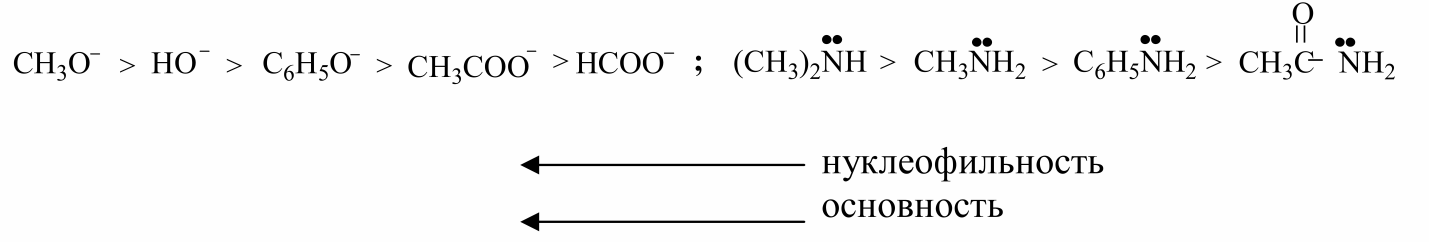
В данных нуклеофилах параллельно основности изменяется и поляризуемость реагента.
Данный параллелизм наблюдается и у реагентов, у которых непо деленная электронная пара находится у элементов, принадлежащих к одному и тому же периоду, но разным группам:

Симбатное изменение нуклеофильной реакционной способности и основности наблюдается не всегда. Так, например, в ряду:
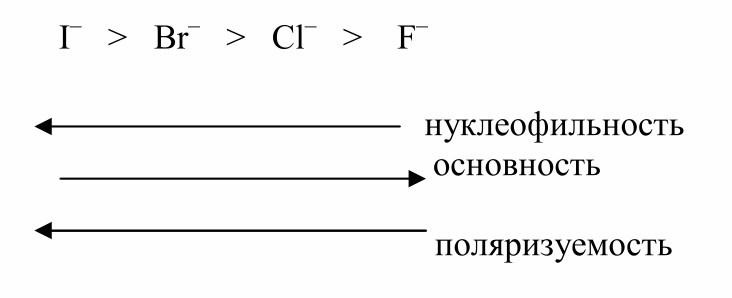
нуклеофильная сила растет, а основность в этом ряду падает.
В данном ряду высокая нуклеофильность связана не с высокой основностью, а с их высокой поляризуемостью.
Найти эксперта


